Home
Welcome to the read2tree wiki!
Read2Tree is a package to infer Multiple Sequence Alignment which could be used to infer thee species tree. The input of R2T is either DNA or RNA sequencing reads.
You can see how different Read2Tree is compared to the standard pipeline of species tree inference in the following figure.
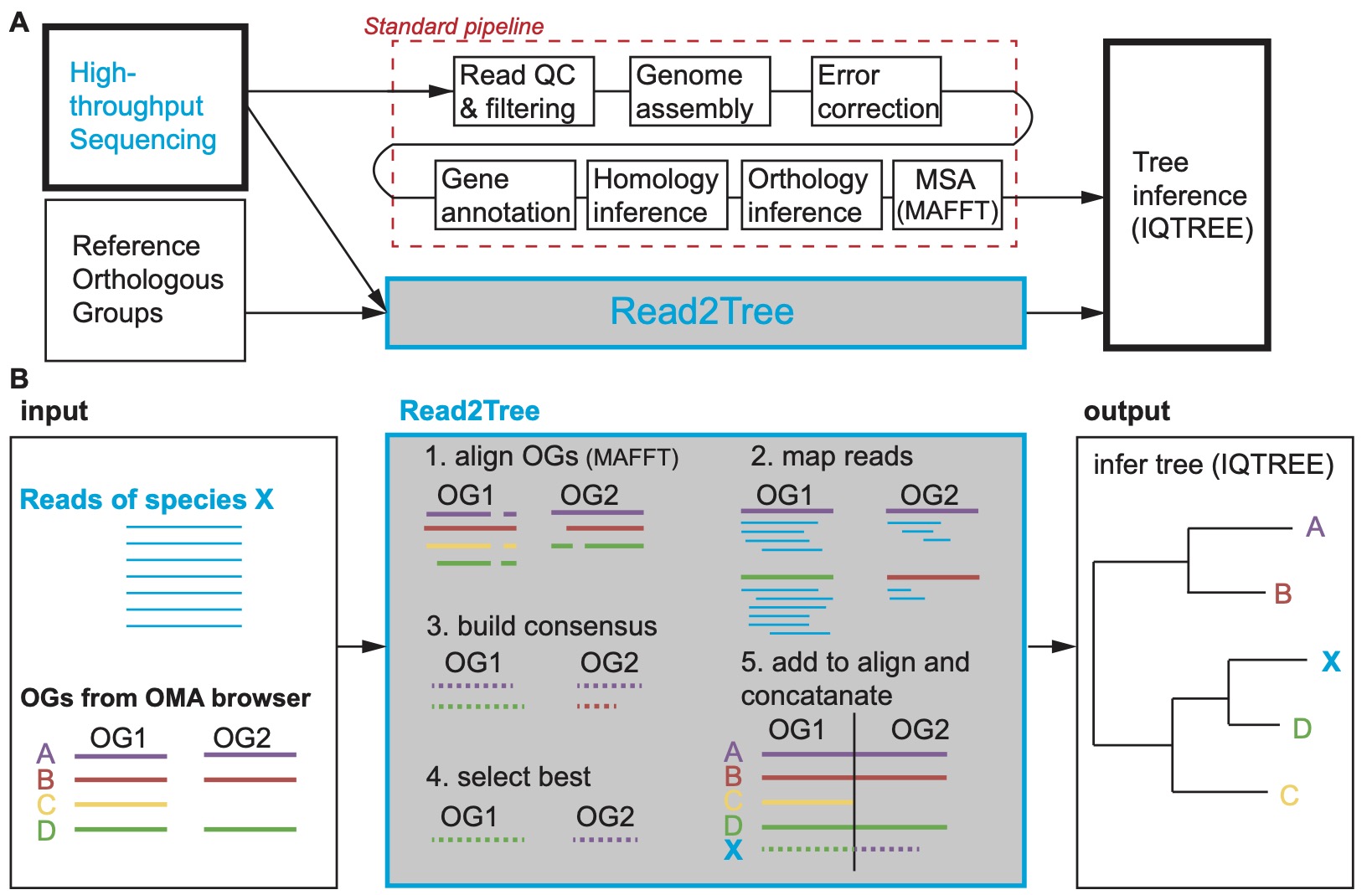
This wiki contains useful information for the read2tree package. Let's get started !
To run read2tree two things are required as input:
1- The DNA sequencing reads as FASTQ file(s). 2- A set of reference orthologous groups, i.e. marker genes.
In the page How to obtain marker genes? we describe how to obtain the latter using OMA browser.
You need to know which argument to use with Read2Tree, which could be found here The details of arguments. For example for your short or long read sequencing data, we use either NGM or NGMLR package as the read aligner.
Once you finish the run, you could the output log of yours with the expected output log.
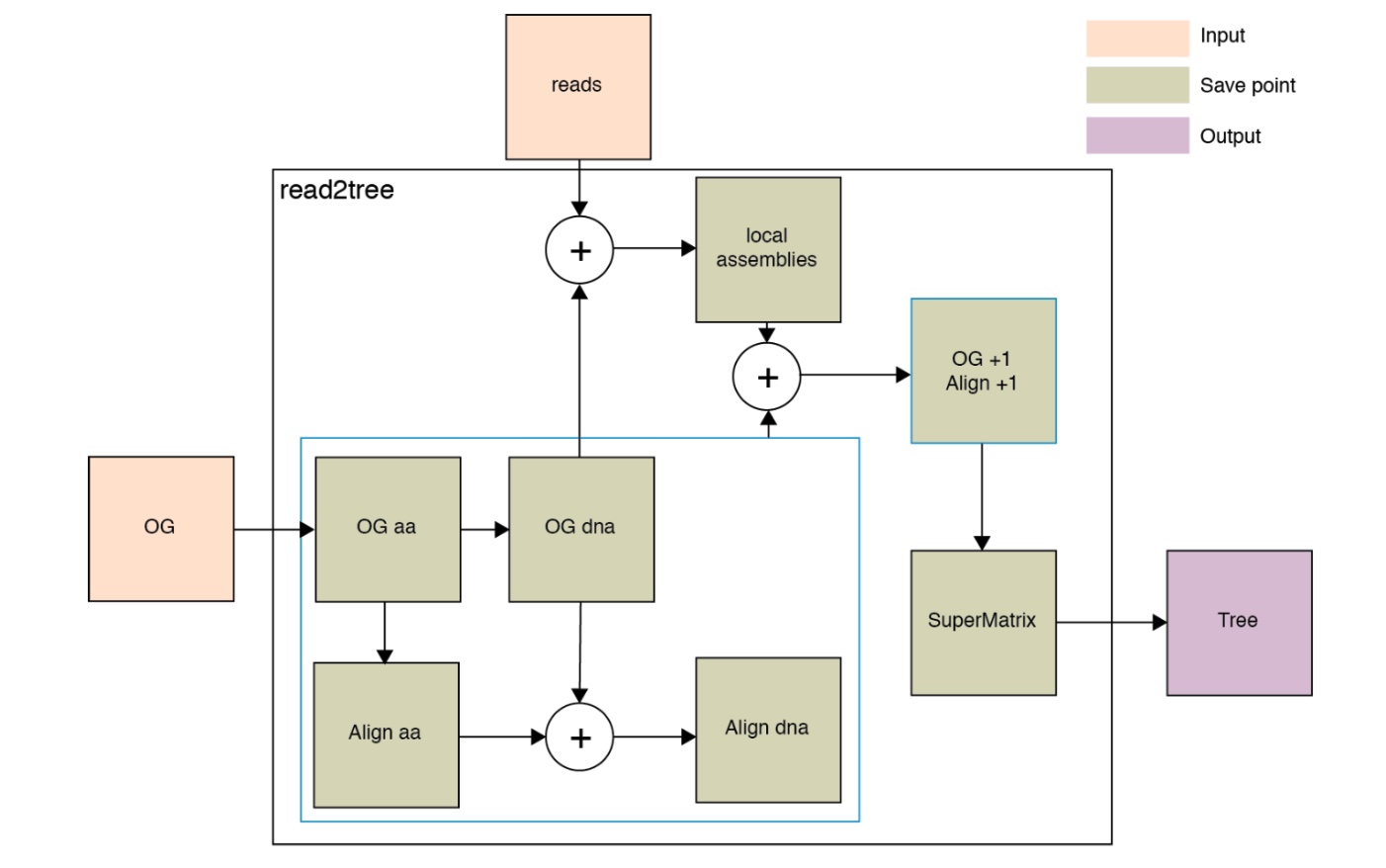
Besides, you could check the intermediate files, the description of output files. This is based on the following figure showing how it works.
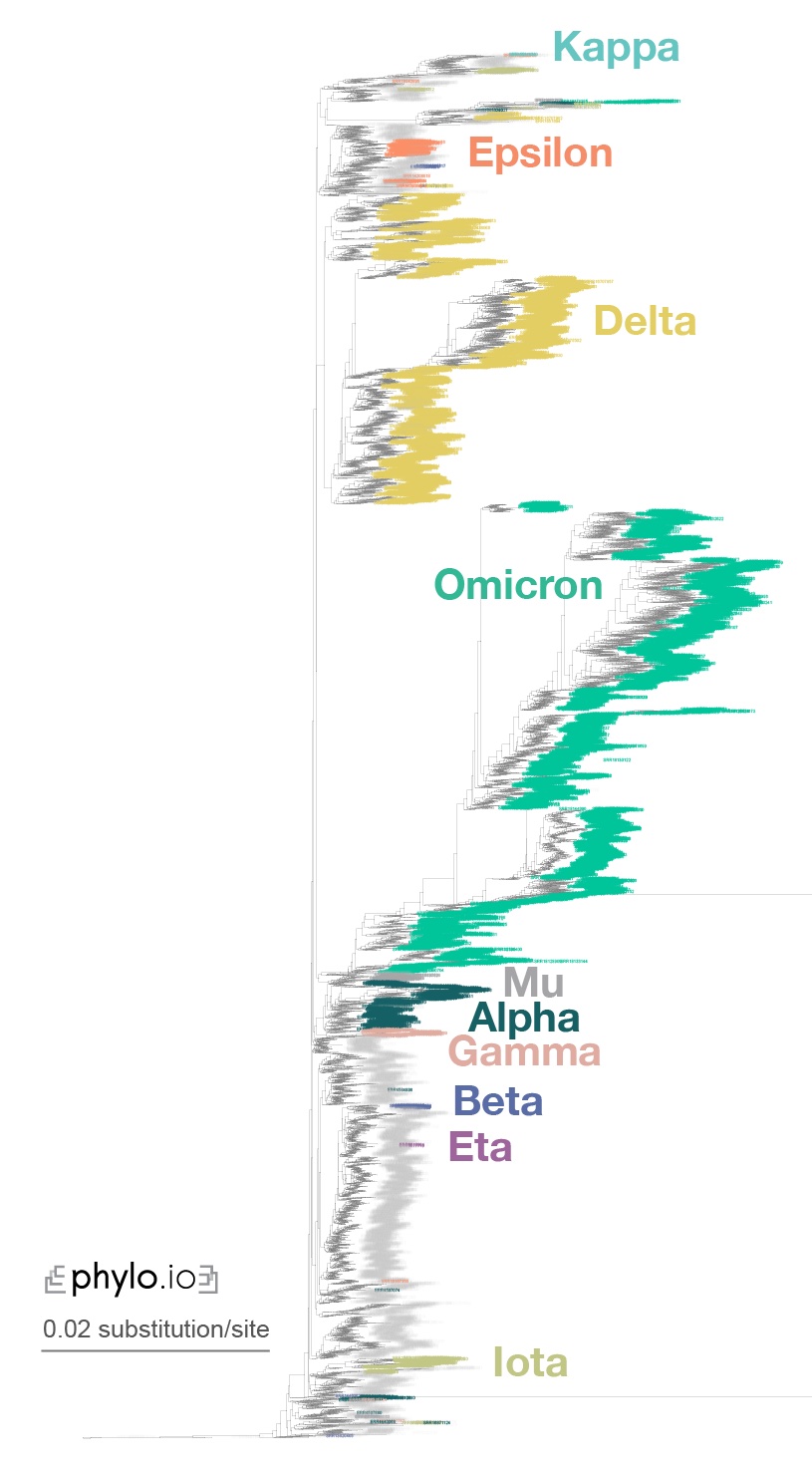
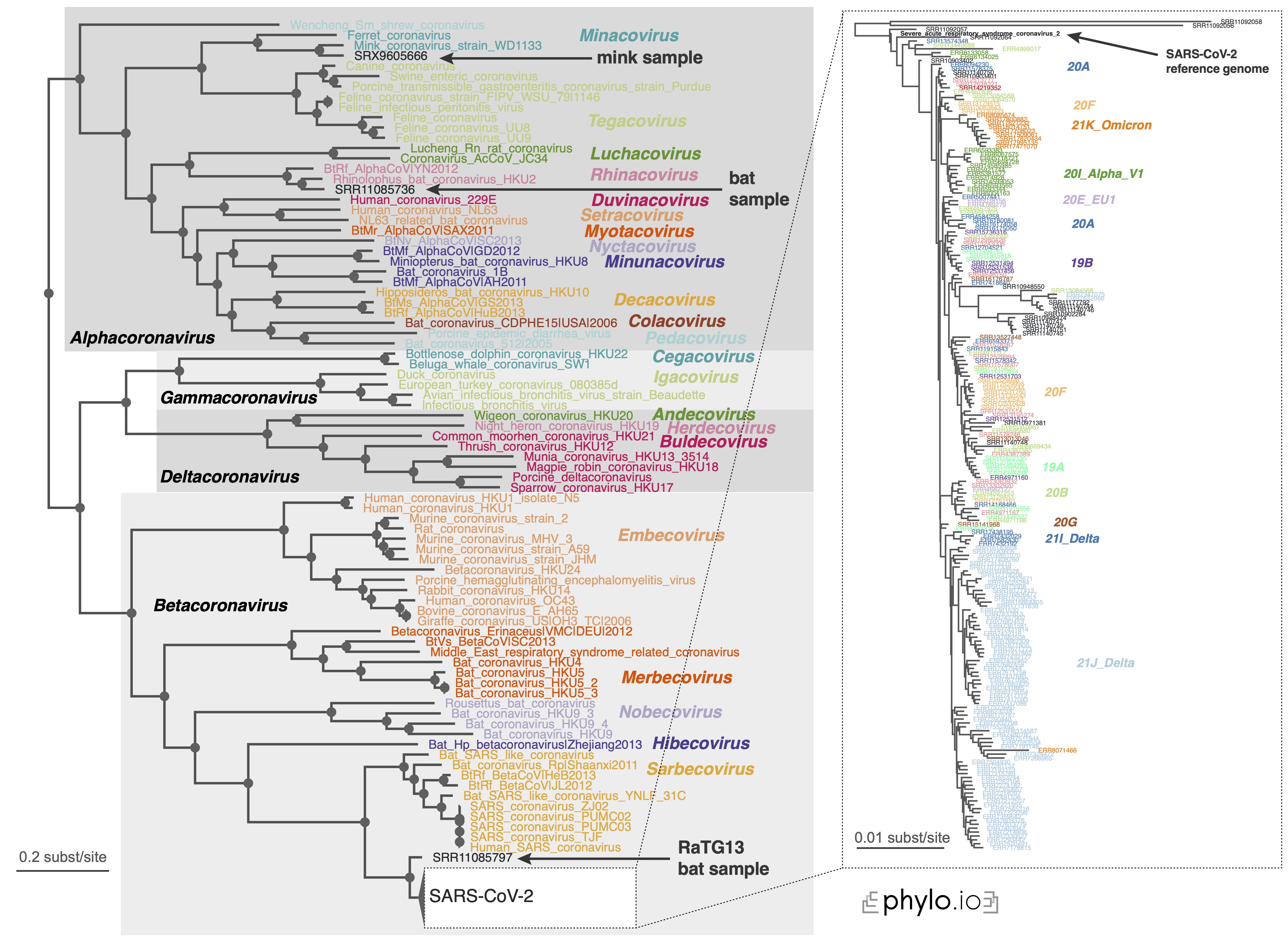
If you want to reproduce the results of the Read2Paper, you may check here. Specifically, for covid analysis, check out this instruction.
We were able to infer the phylogeny for 10,283 covid species within reasonable computation time.
And the final remark is that we actively help users to set up their system to use Read2Tree. Feel free to create a new issue here or contact us by email.