{kind=link}
{kind=link}
{kind=link}
{kind=link}
The project can be used to study the evolution of genome since it is able to find the ancestral states of a large number of genetic variants.
The results provided by this project will give researchers a large number of ancestral states data which helps them with their researches.
ApeShape is not a software, the project is consisted of scripts, and the order of using them is in 'How to use' section.

The data_preparation.R can gather chromosome numbers and SNP postions from NCBI .vcf file. The .bed file generated should be feeded to https://genome.ucsc.edu/cgi-bin/hgCustom. In the website, you should click "Add custom track". The page will be directed to a place where you can upload your file. After your file is uploaded, a page like this:

should appear, and you can select it and choose view in tabe browser.
Now you will enter this page
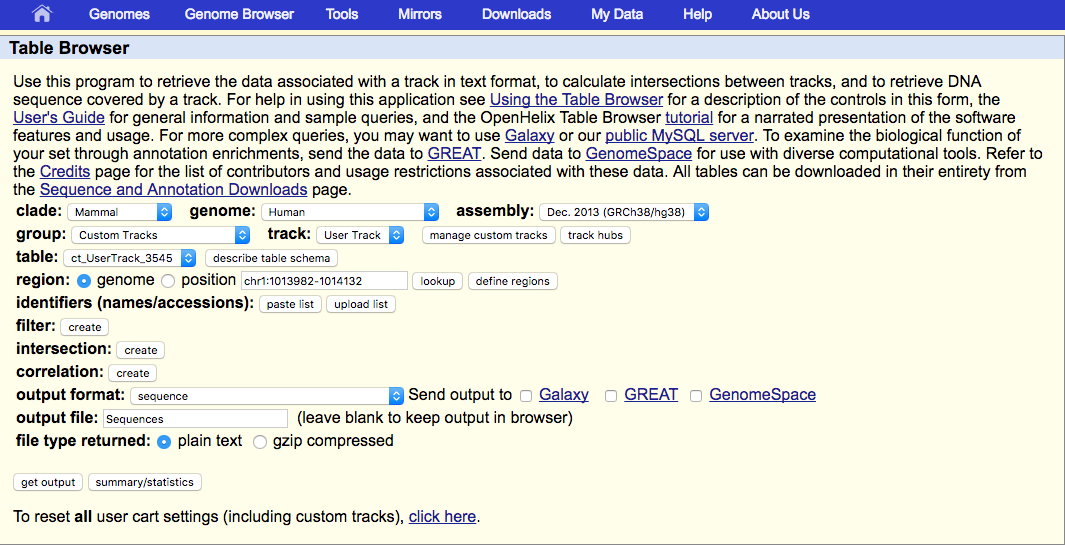
In 'Output format section choose 'sequence', and type in a file name in 'output file' section. Click 'get output' and a file will start to download. This is the sequence file prepared for BLAST.
A custom blast database was created using the reference genomes of Human, Gorilla, Chimpanzee, Orangutan, and Macaque monkey. Using blastn, we found sequences that were similar in all of the chosen organisms. For our analysis, we chose to only keep homologs, however a future analysis could involve the use of paralogs.
Clustal was used to align the sequences that were found during the blast search. This was done to allow for the creation of the phylogeny tree in the next step
The R package Phangorn was used to determine the ancestral allele from the now alligned sequences
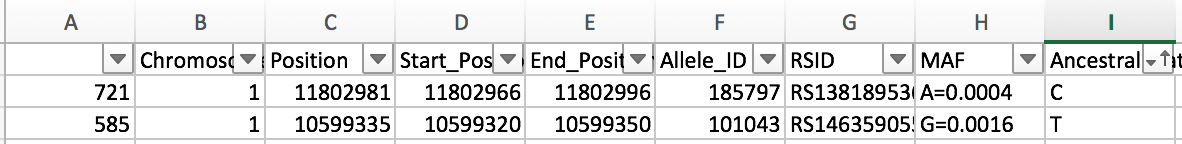
Here is a sample result of the workflow.